Chapter 2 Introduction to Statistical Modeling and Designed Experiments
We begin our dive into Statistical Modeling by building from some introductory statistics material, namely two-sample inference and analysis of variance. The learning objective of this unit include:
- Distinguishing between observational studies and designed experiments.
- Understanding the basic structure of statistical model.
- Understanding that statistical inference can be formulated as the comparison of models.
- Performing a full analysis involving a one-factor experiment.
2.1 Observational Studies versus designed experiments
Before we introduce some details on statistical models we need to step back and discuss the source of our data. Are the data a “sample of convenience”, or were they obtained via a designed experiment or some random sampling scheme or by some sort of survey? How the data were collected has a crucial impact on what conclusions can be meaningfully made.
It is important to recognize that there are two primary methods for obtaining data for analysis: designed experiments and observational studies. There is a third type of data collected through survey sampling methods that incorporates elements from both designed experiments and observational studies, but are excluded here. It is important to know the distinction between each type of data, as this results in a different approach to interpreting estimated models.
2.1.1 Observational Studies
In many situations a practitioner (perhaps you, or a client) simply collect measurements on predictor and response variables as they naturally occur, without intervention from the data collector. Such data is called observational data.
Interpreting models built on observational data can be more challenging. There are many opportunities for error and any conclusions will carry with them substantial unquantifiable uncertainty. Nevertheless, there are many important questions for which only observational data will ever be available. For example, how else would we study something like differences in prevalence of obesity, diabetes and other cardiovascular risk factors between different ethnic groups? Or the effect of socio-economic status on self esteem? It may be impossible to design experiments to investigate these questions, so we must make the attempt to build good models with observational data in spite of their shortcomings.
In observational studies, establishing causal connections between response and predictor variables is nearly impossible. In the limited scope of a single study, the best one can hope is to establish associations between predictor variables and response variables. But even this can be difficult due to the uncontrolled nature of observational data. Unmeasured and possibly unsuspected “lurking” variables may be the real cause of an observed relationship between \(Y\) and some predictor \(X\).
In observational studies, it is important to adjust for the effects of possible confounding variables. Unfortunately, one can never be sure that the all relevant confounding variables have been identified. As a result, one must take care in interpreting estimated models involving observational data.
For the next few chapters we will concentrate on analyses of Designed experiments but the topic of observational studies will be more thoroughly visited later.
2.1.2 Designed experiments
In a designed experiment, the researcher has control over the settings of the predictor variables. For example, suppose we wish to study several physical exercise regimens and how they impact calorie burn where we (the experimenter) assign the different exercise regimes to participants. Typically the values of the predictor variables are discrete (that is, a countably finite number of controlled values). Because of this, predictor variables of this type are known as factors. The experimental units (EUs) are the people we use for the study. We can control some of the predictors variables in this study, such as the amount of time spent exercising or the amount of carbohydrates consumed prior to exercising. Some other predictors may not be controlled but could be measured, such as baseline metabolic variables and the partipant’s age. Other variables, such as the temperature in the room or the type of exercise done, could be held fixed.
Having control over the conditions in an experiment allows us to make stronger conclusions from the analysis. Another key feature from a designed experiment is that the model is chosen for us! We will see later in the class that with observational data we typically need to select a model, but with a designed experiment the model is predetermined based on the experiment.
2.2 Designed experiement vocabulary
2.2.1 What is an experiment?
In an experiment, a researcher manipulates one or more variables, while holding all other variables constant. The main advantage of well-designed experiments over observational studies is that we can establish cause and effect relationships between the predictors and response.
One of the most important things to keep in mind with analyzing designed experiments is that the structure of the experiment dictates how the analysis may proceed. There are a plethora of structural possibilities of which we will only scratch the surface here.
Consider, for example, these four scenarios:
Suppose a study is conducted on the effects a new drug has on patient blood pressure. At the start of the study the blood pressure of all participants is measured. After receiving the drug for two months the blood pressure of each patient is measured again. The analysis to determine the effect of the drug on blood pressure is the Paired \(t\)-test from your introductory statistics class. Here the pairing of each observation is the difference between the before and after blood pressure.
Consider a simple design with a single categorical input variable, or one factor, where each subject (experimental unit) is measured only under one of the factor levels. For example, suppose three different drugs are administered to 18 subjects. Each person is randomly assigned to receive only one of the three drugs. A response is measured after drug administration. The analysis we use for such data is known as a one-way analysis of variance.
We may have a design with two factors, where every level of the first factor appears with every level of the second factor. For example, suppose your data are from an experiment in which alertness levels of subjects were measured after they had been given one of three possible dosages of a drug and whether or not they were allowed to nap for 20 minutes. Such an experimental design is known as a factorial design. Here, the two factors are drug dosage and the occurrence of a nap. The analysis we use for such data is known as a two-way analysis of variance.
Suppose we have a design with only one factor of interest (word type). Five subjects are asked to memorize a list of words. The words on this list are of three types: positive words, negative words and neutral words. The response variable is the number of words recalled by word type, with a goal of determining if the ability to recall words is affected by the word type. Note that even though there is only one factor of interest (word type) with three levels (negative, neutral and positive), this data structure differs greatly from a one-way analysis of variance mentioned above because each subject is observed under every factor level. Thus, there will be variability in the responses within subjects as well as between subjects. This fact will affect how we analyze the data. This is known as a within-subjects analysis of variance, or repeated measures.
We will cover each of these analyses.
2.2.2 Analysis of variance
Analysis of variance (ANOVA) is an analytic tool in statistics to partition the variation in a response variable and attribute it to known sources in the experiment. In practice, it is essentially an extension of the more familiar \(t\)-test. As stated earlier, there are many different experimental structures possible when conducting an experiment and ANOVA can adapt to many of those structures. Some of the other widely used structures in practice are known as nested designs, split-plot designs, and repeated measures designs. It is worth reiterating that the structure of the experimental data will dictate how an analysis of variance is used to model the data. In each of these designs, some variant of an Analysis of Variance is used for analysis.
2.2.3 Elements of a designed experiment
All experiments have factors, a response variable, and experimental units. Here are some definitions and terms:
Factors: A factor is an explanatory variable manipulated by the experimenter. Each factor has two or more levels (i.e., different values of the factor). Combinations of factor levels form what are called treatments. The table below shows factors, factor levels, and treatments for a hypothetical experiment:
Vitamin E
0 mg
250 mg
500 mg
0 mg
Treatment 1
Treatment 2
Treatment 3
400 mg
Treatment 4
Treatment 5
Treatment 6
In this hypothetical experiment, the researcher is studying the possible effects of Vitamin C and Vitamin E on health. There are two factors: dosage of Vitamin C and dosage of Vitamin E. The Vitamin C factor has three levels: 0 mg per day, 250 mg per day, and 500 mg per day. The Vitamin E factor has 2 levels: 0 mg per day and 400 mg per day. The experiment has six treatments. Treatment 1 is 0 mg of E and 0 mg of C; Treatment 2 is 0 mg of E and 250 mg of C; and so on.
Response variable: In the hypothetical experiment above, the researcher is looking at the effect of vitamins on health. The response variable in this experiment would be some measure of health (annual doctor bills, number of colds caught in a year, number of days hospitalized, etc.).
Experimental units: The recipients of experimental treatments are called experimental units. The experimental units in an experiment could be anything - people, plants, animals, or even inanimate objects.
In the hypothetical experiment above, the experimental units would probably be people (or lab animals). But in an experiment to measure the tensile strength of string, the experimental units might be pieces of string. When the experimental units are people, they are often called participants; when the experimental units are animals, they are often called subjects.
Three characteristics of a well-designed experiment. While the elements above are common to all experiments, there are aspects to the manner in which the experiment is run which are critical to it being a “well-designed” experiment. A well-designed experiment includes design features that allow researchers to eliminate extraneous variables as an explanation for the observed relationship between the factor(s) and the response variable. Some of these features are listed below.
Control: Control refers to steps taken to reduce the effects of extraneous variables (i.e., variables other than the factor(s) and response). These extraneous variables are called lurking variables. Control involves making the experiment as similar as possible for experimental units in each treatment condition. Two control strategies are control groups and placebos:
Control group: A control group is a baseline group that receives no treatment or a neutral treatment. To assess treatment effects, the experimenter compares results in the treatment group to results in the control group.
Placebo: Often, participants in an experiment respond differently after they receive a treatment, even if the treatment is neutral. A neutral treatment that has no “real” effect on the dependent variable is called a placebo, and a participant’s positive response to a placebo is called the placebo effect. To control for the placebo effect, researchers often administer a neutral treatment (i.e., a placebo) to the control group. The classic example is using a sugar pill in drug research. The drug is effective only if participants who receive the drug have better outcomes than participants who receive the sugar pill.
Blinding: Of course, if participants in the control group know that they are receiving a placebo, the placebo effect will be reduced or eliminated; and the placebo will not serve its intended control purpose. Blinding is the practice of not telling participants whether they are receiving a placebo or even what treatment they receive. In this way, participants in the control and treatment groups experience the placebo effect equally. Often, knowledge of which groups receive placebos is also kept from people who administer or evaluate the experiment. This practice is called double blinding. It prevents the experimenter from “spilling the beans” to participants through subtle cues; and it assures that the analyst’s evaluation is not tainted by awareness of actual treatment conditions.
Randomization: Randomization refers to the practice of using chance methods (random number generation, etc.) to assign experimental units to treatments. In this way, the potential effects of lurking variables are distributed at chance levels (hopefully roughly evenly) across treatment conditions. For example, suppose we had 12 experimental units which we wanted to completely randomly allocate to three different treatments (call them A, B and C). We can accomplish this as follows in R:
ids <- 1:12
trt <- rep(c('A','B','C'), each=4)
randomized <- sample(ids, size=length(ids), replace=FALSE)
CRD <- data.frame(trt, randomized)
CRD## trt randomized
## 1 A 9
## 2 A 3
## 3 A 11
## 4 A 2
## 5 B 6
## 6 B 1
## 7 B 8
## 8 B 12
## 9 C 10
## 10 C 7
## 11 C 5
## 12 C 4Such a design is known as a completely randomized design (or CRD).
Randomization is arguably one of the most important pieces of any well-designed experiment. Failure to randomize can lead to confounded conclusions (see discussion below) and biased results!
Example: Consider a contrived, but simple, experiment where you might be comparing a new heart disease drug to a placebo. Suppose your sample consists of one-hundred adults with with diagnosed heart disease. Further, suppose the ages of the adults are uniformly distributed across the range 20 to 90. Out of convenience suppose the new drug is administered to the youngest fifty volunteers and the placebo to the oldest 50 volunteers. Would you trust the results of this experiment?
However, if the drug/placebo assignment was randomly assigned to different patients of different ages and backgrounds, the randomization would help eliminate any bias age.
Replication: Replication refers to the practice of assigning each treatment to many experimental units. The purpose of replication is as follows:
- Estimate the noise in the system. Recall earlier in the text when we discussed sources of variation in data. If you were to apply precisely the same treatment to a set of different individuals, the response would naturally vary due to myriad reasons of little to no interest to the researcher. So, responses vary to some degree due to simple random chance fluctuations, or “noise.” For our purposes, this constitutes a single source of variation.
- Now, lets apply different treatments to a set of individuals. The responses would still vary, but now there are two sources of variation: the random chance element from above (because we still have different individuals), but now we also include that the different treatments may have different effects on the response. So, one purpose of replication is so that we can estimate how much noise is naturally in the system, and can see if the variability in response introduced by changing the treatment rises above the noise. The more replication in an experiment, the better you can estimate the noise. However, this comes at an expense!
- Help wash out the effect of lurking variables. This was also mentioned as a reason for randomization. When coupled with randomization, replication provides the “opportunity” for your experiment to have a mix of all those lurkers appear under each treatment, thus mitigating their effects from biasing your results.
2.2.3.1 Confounding
This sounds serious but what is it?
Confounding is a condition that occurs when the experimental controls do not allow the experimenter to reasonably eliminate plausible alternative explanations for an observed relationship between factors and the response. Needless to say, confounding renders experimental results to be seriously impaired, if not totally useless. Consider this example:
Example: A drug manufacturer tests a new cold medicine with 200 participants: 100 men and 100 women. The men receive the drug, and the women do not. At the end of the test period, the men report fewer colds.
What is the problem? This experiment implements no controls! As a result, many variables are confounded, and it is impossible to say whether the drug was effective. For example:
- Biological sex is confounded with drug use. Perhaps, men are less vulnerable to the particular cold virus circulating during the experiment, and the new medicine had no effect at all. Or perhaps the men experienced a placebo effect.
This experiment could be strengthened with a few controls. Women and men could be randomly assigned to treatments. One treatment group could receive a placebo. Blinding could be implemented to reduce the influence of “expectation” on the part of participants with regard to the outcome of a treatment. Then, after all this, if the treatment group (i.e., the group getting the medicine) exhibits sufficiently fewer colds than the control group, it would be reasonable to conclude that the medicine was the reason for effectively preventing colds, and not some other lurking reason.
2.3 Statistical Analyses is Modeling
Think of a model airplane or a Lego kit of the Eiffel Tower – it is just a simplified representation of the real thing.
In many statistical problems (and all the problems encountered in this course), we are interested in describing the relationship that may exist among several different measured variables. For example, your current GPA could be impacted by how much you study. And there may be several contributing factors – your credit hour load, your ACT score, whether or not you have a part-time job, etc.
Of course, what makes your GPA is the result of a very highly complex combination of factors, some important and others not so important.
A statistical model is a mechanism to describe the structural relationship between some measured outcome (called the response variable) and one or more impacting variables (called predictor variables or factors or features, depending on the contextual circumstance) in a simplified mathematical way.
It is in this sense that you can think of a statistical model as a “simplification”: we know that the true relationship between response and predictor variables is very detailed and complex. The goal of the model is not to capture all the intricacies of the complexity but rather, the model only seeks to describe the essential features of any relationships that exist.
2.3.1 Example of a two-sample \(t\)-test as a model
It turns out, we have already fit a statistical model. Consider a variation of the two-sample \(t\)-test we performed in the previous chapter on the University Admissions data (Kutner et al., 2004). For cohesiveness we input the data again.
uadata.trim <- uadata |>
filter(year %in% c(1996, 2000) )
t.test(act ~ year, data=uadata.trim, conf.level=0.98,
alternative="two.sided", var.equal=TRUE)##
## Two Sample t-test
##
## data: act by year
## t = 0.3013, df = 277, p-value = 0.7634
## alternative hypothesis: true difference in means between group 1996 and group 2000 is not equal to 0
## 98 percent confidence interval:
## -0.9160716 1.1868643
## sample estimates:
## mean in group 1996 mean in group 2000
## 24.69014 24.55474Looking at the code you’ll note here we are performing a two.sided or “not equal to” test (in chapter 1 we performed a “>” test).
More importantly note the act ~ year notation – this is known as a formula in R.
In particular, we are telling R that the ACT scores are a function of the student’s incoming Year.
This concept should be familiar as it is similar to the regression ideas from your Intro Statistics course.
It turns out that nearly all statistical inference can be framed in terms of modeling. To see this we first must cover some light math.
Recall from Intro Statistics that a random variable \(Y\) is typically assumed to come from a Normal distribution with mean \(\mu\) and standard deviation \(\sigma\). Standard shorthand notation for this is to write \[Y \sim N(\mu, \sigma).\] You may also remember that if you subtract off the mean, \(\mu\), from a random variable it will just shift the distribution by \(\mu\) units. For example \[ Y - \mu \sim N(0, \sigma)\] We could reformulate this equation further with \[\begin{equation} Y = \mu + \varepsilon \tag{2.1} \end{equation}\] where \[\varepsilon \sim N(0, \sigma).\]
The equation (2.1) can be considered a baseline model. Here, we are simply stating that the random variable \(Y\) is a function of a mean \(\mu\) and some random component \(\varepsilon\), which happens to be from a Normal distribution with mean 0 and standard deviation \(\sigma\). In words have the model \[\textbf{Data} = \textbf{Systematic Structure} + \textbf{Random Variation}\] where in the base model the Systematic Structure is simply the constant \(\mu\).
Estimation
A quick note about estimation. In your Intro Stat class you worked with this model regularly. We would estimate \(\mu\) with the sample mean \(\bar{Y}\) and estimate \(\sigma\) with the sample standard deviation \(S\). One-sample hypothesis testing and confidence intervals are all formulated using this baseline model.
Two-sample \(t\)-test as a Model
Now consider the case of a two-sample \(t\)-test; for example, testing if ACT scores were different in 1996 compared to 2000. Let’s create a new variable called \(\tau\) that measures how the 2000 ACT scores deviate from the 1996 ACT scores. That is, consider a model \[Y_i = \mu + \tau + \varepsilon_i\] Here the value \(\mu\) corresponds to the mean ACT score in 1996 and is the baseline, \(\tau\) measures how the year 2000 ACT scores differ from 1996 (increase? decrease? no change?) and \(\varepsilon_i\) is the underlying random part of the \(i^\mathrm{th}\) observation \(Y_i\). Thus, if \(Y_i\) is an observation from the year 2000, the structural component will be \(\mu+\tau\), whereas if the observation is from 1996, the structural component is simply \(\mu\).
Another way to frame this model is to augment with a random variable \(X_i\). If observation \(i\) is from 1996 then \(X_i\) takes on the value zero and if the observation is from 2000 then \(X_i\) takes on the value one. We’ll let \(Y_i\) be the observed ACT score for student \(i\) and consider the following
\[\begin{equation} Y_i = \mu + \delta\cdot X_i + \varepsilon_i. \tag{2.2} \end{equation}\]
Now \(\mu\) is the mean of 1996 ACT scores (when \(X_i=0\)), \(\delta\) is the effect the year 2000 has on mean ACT scores (e.g., suppose mean ACT scores in 1996 were 27, but in 2000 they were 31, thus \(\delta=4\)), thus \(\mu+\delta\) is the mean ACT score in the year 2000 (much like the \(\tau\) effect above). Lastly, \(\varepsilon_i\) is how observation \(i\) deviates from the structural component and we assume \(\varepsilon_i \sim N(0, \sigma)\) for standard deviation \(\sigma\) for all \(i=1,\ldots,n\).
When performing a two-sample \(t\)-test we are essentially testing \(H_0:\ \delta=0\) versus \(H_A:\ \delta\neq 0\). Note that if \(H_0\) were true, we are back to the baseline model (2.1).
So to frame this another way, a two-sample \(t\)-test essentially is a choice of
\[H_0: \textrm{Baseline Model} ~~~ Y_i = \mu + \varepsilon_i\] and \[H_A: \textrm{More complex model} ~~~ Y_i = \mu + \delta\cdot X_i + \varepsilon_i.\]
To tie everything together note that in \(H_A\) we are essentially describing observation \(Y_i\) as a function of the variable \(X_i\). Because of this we typically refer to \(Y_i\) as the response (or dependent) variable and \(X_i\) as a predictor (or independent) variable.
This coincides with the R formula notation, response ~ predictor.
Back to the example
In this observational study, we compared the ACT scores of incoming freshmen in 1996 and compared them to ACT scores for the incoming class of 2000. With a \(p\)-value of 0.7634 on 277 degrees of freedom, we do not have evidence to suggest that the ACT scores for freshmen in 1996 differs from those in 2000.
2.4 One-Way ANOVA
When there is a single factor variable whose levels may only change between different experimental units, we can analyze the effect of the factor on the response using a one-way analysis of variance (one-way ANOVA) on the between-subjects factor. If we had a single factor with just two levels, you could still use a one-way ANOVA, but it would be equivalent to running an independent samples \(t\)-test (see above for an example). In this sense, one-way ANOVA is an extension of the two-sample \(t\)-test (think a \(k\)-sample test).
In this design, we observe random samples from \(k\) different populations. As a designed experiment, these populations may be defined on the basis of an administered treatment. The levels of the factor being manipulated by the researcher form the different treatments. Frequently, the data are obtained by collecting a random sample of individuals to participate in the study, and then randomly allocating a single treatment to each of the study participants. If so, then the individual subjects (experimental units) receiving treatment \(j\) may be thought of as a random sample from the population of all individuals who could be administered treatment \(j\).
The one-way data structure looks like the following:
\[\begin{array}{cccc} \hline \textbf{Treatment 1} & \textbf{Treatment 2} & \cdots & \textbf{Treatment}~k \\ \hline Y_{11} & Y_{21} & \ldots & Y_{k1} \\ Y_{12} & Y_{22} & \ldots & Y_{k2} \\ Y_{13} & Y_{23} & \ldots & Y_{k3} \\ \vdots & \vdots & \vdots & \vdots \\ Y_{1n_{1}} & Y_{2n_{2}} & \ldots & Y_{kn_{k}} \\ \hline \end{array}\] where \(Y_{ji}\) is the \(i^\mathrm{th}\) observation within treatment \(j\), for \(j=1,\ldots,k\) and \(i=1,\ldots, n_j\).
The model for such data has the form \[\begin{equation} Y_{ji} = \mu_j + \varepsilon_{ji} \tag{2.3} \end{equation}\] where \(\mu_j\) is the mean of the \(j^\mathrm{th}\) treatment, \(j=1,\ldots,k\), or more commonly, \[ Y_{ji} = \mu + \tau_j + \varepsilon_{ji} \] where
- \(Y_{ji}\) is the \(i^\mathrm{th}\) observation in the \(j^\mathrm{th}\) treatment.
- \(\mu\) is the overall mean of all the populations combined.
- \(\tau_j\) is the deviation of the mean by the \(j^\mathrm{th}\) treatment population from the overall mean \(\mu\).
- \(\varepsilon_{ji}\) is the random error term.
Similar to the two-sample \(t\)-test formulas above, this equation can also be written as \[ Y_{i} = \mu_1 + \delta_2 X_{2i} + \delta_3 X_{3i} + \ldots + \delta_k X_{ki} + \varepsilon_{i} \] where * \(Y_i\) is the \(i^\mathrm{th}\) observation. * \(\mu_1\) is the mean of the first treatment. * \(\delta_j\) is the different between the \(j^\mathrm{th}\) treatment mean and the first treatment mean; i.e., _j = _1 + _j$. * \(X_{ji}\) is an indicator variable, indicating if the \(i^\mathrm{th}\) observation is in the \(j^\mathrm{th}\) treatment; i.e., \(X_{ji}=1\) when the \(i^\mathrm{th}\) observation is in group \(j\), otherwise \(X_{ji}=0\).
The usual test of interest in a one-way analysis of variance is to compare the population means. The null and alternative hypotheses are given by:
\[H_0: \mu_1 = \mu_2 = \ldots = \mu_k\] \[H_a: ~\textrm{at least two of the population means differ}\]
Expressed in terms of the more commonly used model parameters, we really want to test to see if there are no treatment mean deviations among the populations, so the above hypotheses can be equivalently expressed as follows:
\[H_0: \tau_1 = \tau_2 = \ldots = \tau_k = 0\] \[H_a: ~\text{at least one } \tau_j \neq 0~~~~~\text{for}~j=1,\ldots, k\]
An alternative formulation is based on the coefficients on the indicator functions:
\[H_0: \delta_2 = \delta_3 = \ldots = \delta_k = 0\] \[H_A: ~\text{at least one } \delta_j \neq 0~~~~\text{for}~j=2,\ldots,k\] With this last formulation, under \(H_0\) all the \(\delta_j\) terms equal zero, so the \(X_{ji}\) terms have no effect, and the model reduces to the baseline model \(Y = \mu+\varepsilon\).
In terms of modeling, we are testing”
\[H_0: \textrm{Baseline Model:} ~~ Y_{ji} = \mu + \varepsilon_{ji}\] \[H_a: \textrm{More Complex Model:} ~~ Y_{ji} = \mu + \tau_j + \varepsilon_{ji},~~\textrm{at least one}~\tau_j\neq0\] and we note the more complicated model can be expressed a few different ways.
We test these equivalent hypotheses by partitioning the total variability in the response into two components: (1) variation between treatments, and (2) variation within treatments. The former, (1) variation between treatments, effectively is the variability that is explained by the structural components in the model. The latter partition is essentially residual error, or unexplained variability. The details of this partitioning are outside the scope of this text, but we will hint at the ideas further in Chapter 6. The resulting method for this hypothesis is the ANOVA \(F\)-test.
The \(F\)-test works like other hypothesis test you may have seen (e.g., the \(t\)-test or \(\chi^2\) goodness-of-fit test). The test statistic is a ratio of variance estimates and follows the Fisher-Snedecor distribution (or \(F\)-distribution for short). The details of this distribution are not important for this course and are covered in a Mathematical Statistics course. An example of a one-way ANOVA in R is provided below.
2.4.1 Example of One-Way ANOVA
Example: Drug effects on “knockdown” times (from the text Walpole et al. (2007)).
Immobilization of free-ranging white-tailed deer by drugs allows researchers the opportunity to closely examine deer and gather valuable physiological information. Wildlife biologists tested the “knockdown” time (time, measured in minutes, from injection to immobilization) of three different immobilizing drugs. Thirty male white-tailed deer were randomly assigned to each of three treatments: Group A received 5mg of liquid succinylcholine chloride (SCC); group B received 8 mg of powdered SSC; and group C received 200 mg of phencyclidine hydrochloride.
Original Source: Wesson (1976)
The data appear in the R dataframe deerKnockdown.RData in our repository: (https://tjfisher19.github.io/introStatModeling/data/deerKnockdown.RData)
site <- "https://tjfisher19.github.io/introStatModeling/data/deerKnockdown.RData"
load(url(site))
glimpse(deer_knockdown_times)## Rows: 30
## Columns: 2
## $ Drug <chr> "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "B", …
## $ Knockdown_time <dbl> 11, 5, 14, 7, 10, 7, 23, 4, 11, 11, 10, 7, 16, 7, 7, 5,…Note the subtle change in the code compared to read_csv() and read_table(). When loading an R data.frame from a website, we need to use the url() function to open and close the internet connection inside the load() function.
We can then plot the data.
ggplot(deer_knockdown_times, aes(x=Knockdown_time, y=Drug)) +
geom_boxplot() +
geom_jitter(height=0.25) +
stat_summary(fun="mean", geom="point", shape=23, fill="gray60", size=4) +
labs(x="Time to Immobilization (min)", y="Drug Treatment" ) +
theme_minimal()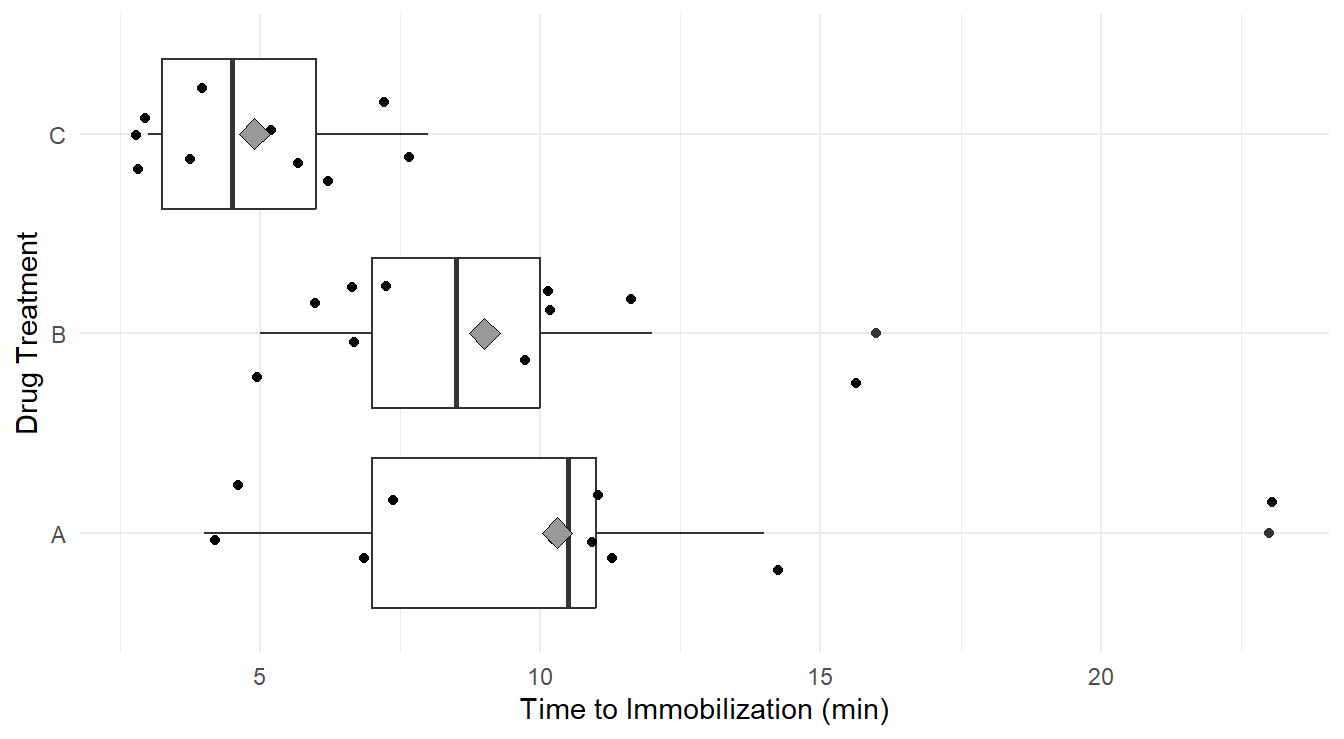
Figure 2.1: Side-by-side Box-whiskers plots, with data overlayed jittered data, and treatement means (gray diamond), showing the distribution of ‘knockdown’ times for each of the three drug treatments, A, B and C. Overall we see immobilization times is highest in treatment A, followed by B and C. We Also note that variability appears greatest in treatment A, followed by B and C.
Visually it appears the immobilization times under drug C seem to be noticeably lower on average than for the other two drugs. Also, we may suspect the variance of immobilization times is different between the three drugs. That is, the distributions of times for drugs A and B are more disperse than for drug C.
To perform One-Way ANOVA we employ the aov function (for Analysis Of Variance).
That’s it! The single line of code above fits the model described in (2.3) with \(k=3\) treatments corresponding to the three Drug types.
Before proceeding with the analysis of the model output, we need to check the assumptions of our underlying model.
2.5 Model Assumptions & Assessment
Arguably the most important aspect in an analysis is the assumptions made with the chosen statistical model. In the field of statistics there are two types of assumptions made:
- Assumptions about the systematic structure.
- Assumptions about the random variation.
Most of the time, when referring to “assumptions” we are concerned about 2: Assumptions about the random variation. However, it is important to consider the assumptions on the structure.
2.5.1 Structural Assumptions
In all of our models/testing discussed above, we are assuming the response variable is a linear function on the predictor variable(s) (consider (2.2) and (2.3), both are linear equations).
This assumption becomes more important later in the course when discussing lintear regression.
Here, since the model is looking at factor, or categorical treatments, as the inputs, the linear equation reduces to comparing the mean of each group.
Based on context, it is important to consider if this structure is appropriate.
2.5.2 Random Variation Assumptions
In this class, along with nearly all methods you learned in your introductory statistics course, we make the following assumptions about the underlying random variation (i.e., stochastic part) of the response variable, \(\varepsilon\):
- The \(\varepsilon_i\) terms are independent.
- The variance of \(\varepsilon_i\) is homogeneous; i.e., \(Var(\epsilon_i)=\sigma^2\) is constant for all \(i=1,\ldots,n\). This is sometimes known as being homoskedastic or having “constant variance”.
- The \(\varepsilon_i\) terms are Normally distributed.
Collectively, the three assumptions simply state \[\varepsilon_i \stackrel{iid}{\sim} N(0, \sigma^2), ~~\textrm{for variance}~\sigma^2\]
Important Note. In the above bullet points, the assumptions are listed in their order of importance. If you lack independence, nearly all statistical methods we cover in this text are invalid. The constant variance assumption is important but in many cases violations of the assumption can be addressed. Lastly, the Normality assumption tends to not be all that important except in small sample sizes and when the data are heavily skewed – but we can also address issues with Normality in many cases.
2.5.2.1 Independence
In general, independence (or lack thereof) is the result of the sampling scheme employed during data collection. If you collect your data according to a well designed experiment or a simple random sampling scheme with one observation per subject, there is usually no reason to suspect you will have a problem. Data collected sequentially in time (e.g., daily high temperatures or repeated measures on the same subject) will exhibit a phenomenon known as autocorrelation (also called serial correlation) and is a major problem! Specialized methods are needed for this type of data! We will briefly cover the idea of repeated measures in Section 4.2, while a general treatment of autocorrelation is excluded from our work.
2.5.2.2 Constant Variance
To assess the constant variance assumption we typically consider two simple plots: the ‘Residuals vs Fitted’ plot and ‘Scale-Location’ plots as seen in Figure 2.2 (code to generate the plot is provided below).
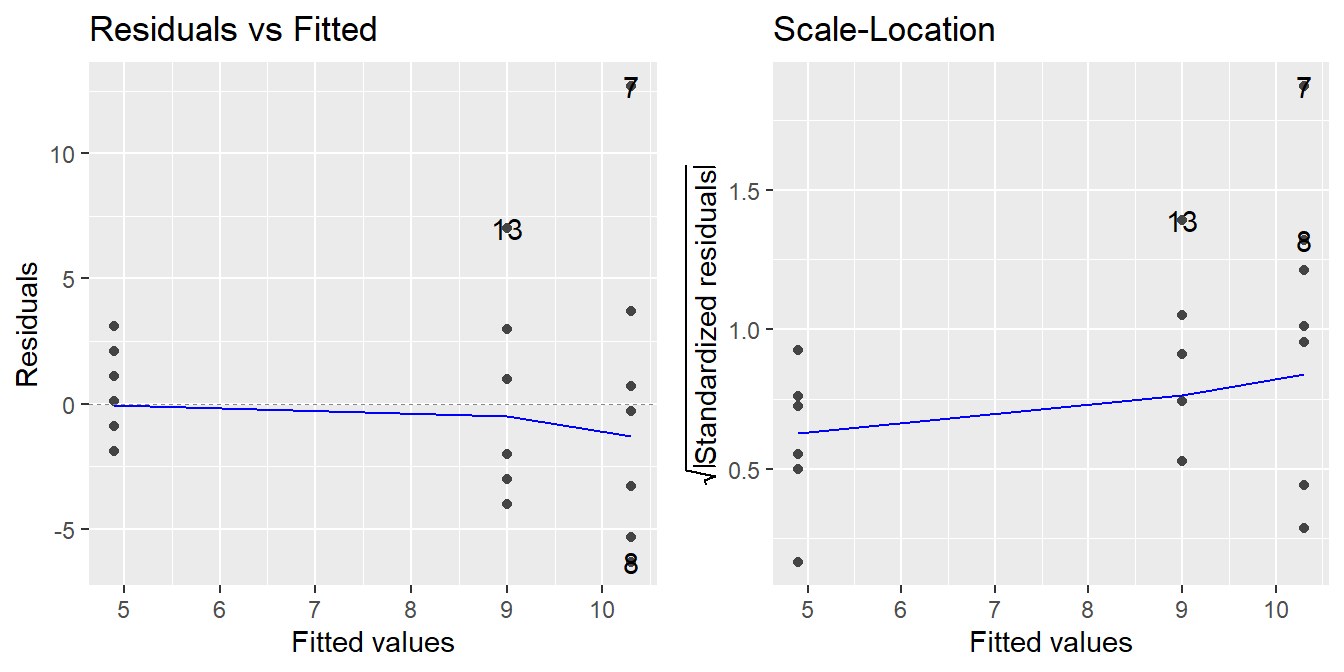
Figure 2.2: Residual diagnostic plots showing a fanning effect in the residuals vs fitted and increasing linear trend in the Scale-Location plot, thus suggesting the variance increases with the mean.
Before reading these plots, we need to answer two questions: What is a residual? and What is a fitted value?
Consider the baseline model (2.1), \(Y = \mu + \varepsilon\). We can estimate \(\mu\) with \(\bar{y}\), then we could predict each unobserved \(Y_i\) with \(\hat{Y}_i=\bar{y}\). From there, we can approximate the random noise of the \(i^\mathrm{th}\) observation \(\varepsilon_i\) with \(e_i = Y_i - \hat{Y}_i = Y_i - \bar{y}\). The values \(\hat{Y}_i\) are known as the fitted, or predicted, values and the \(e_i\) terms are the sample residuals.
In the case of the One-Way ANOVA model (2.3), we would calculate a different mean \(\bar{y}_J\) for each of the \(k\) treatments and each \(Y_{ji}\) would be predicted by the appropriate \(\bar{y}_j\) for \(j=1,\ldots,k\). The sample residuals would be found by \(e_{ji} = Y_{ji} - \hat{Y}_{ji} = Y_{ji} - \bar{y}_j\).
How to read these plots?
A residuals versus fitted plot is provided in the left panel of Figure 2.2. If all is well, you should see fairly uniform (“constant”) spread of the points in the vertical direction and the scatter should be symmetric vertically around zero. The vertical spread here looks like it might be expanding as the fitted values increase (look at the dots from left-to-right, we see a ‘fanning’ effect where the vertical spread for fitted values around 5 is more narrow, than the spread at fitted values of 9, and the largest is at the fitted values near 10.5), suggesting that there may be non-constant error variance.
Note: In the case of ANOVA, you will also see stacked dots, or vertical stratification in the Residuals vs Fitted plot. This is an artifact of the fitted values – all observations in the same treatment are predicted with the same value (\(\bar{y}_j\)), so you should expect to see this stratification. Also, when looking at ANOVA, we can generally ignore the plotted trend line (blue line in the left panel above). It will be more useful when assessing linear regression.
An attempt at refining the residuals vs fitted plot is given in the plot on the right in Figure 2.2, called the Scale-Location plot. The difference now is that instead of plotting the raw residuals \(e_{ij}\) on the vertical axis, R first standardizes them (so you can better check for extreme cases), takes their absolute value (to double the resolution in the plot) and then takes their square root (to remove skew that sometimes affects these plots). R then adds a trend line through the points: if the constant variance assumption is reasonable, this trend line should be roughly horizontal. Here, you can see it trending upward, so we likely have a variance that is increasing with the predicted values (i.e., we not have constant variance).
Note: Using the absolute values of the residuals in the Scale-Location plot is often a good refinement. The reason being we typically do not care if a residual is positive or negative: all we are looking for is if the scatter is uniform across the plot. Thus, by looking at the magnitude of the residuals only (and not their direction), we are actually improving the resolution of the information that the plot contains in addressing constant variance. Also, like with the Residuals vs Fitted plot, we will see vertical stratifications since our fitted values are the sample mean within a treatment group.
2.5.2.3 Checking Normality
We typically make this assessment with a Normal quantile-quantile (Q-Q) plot.
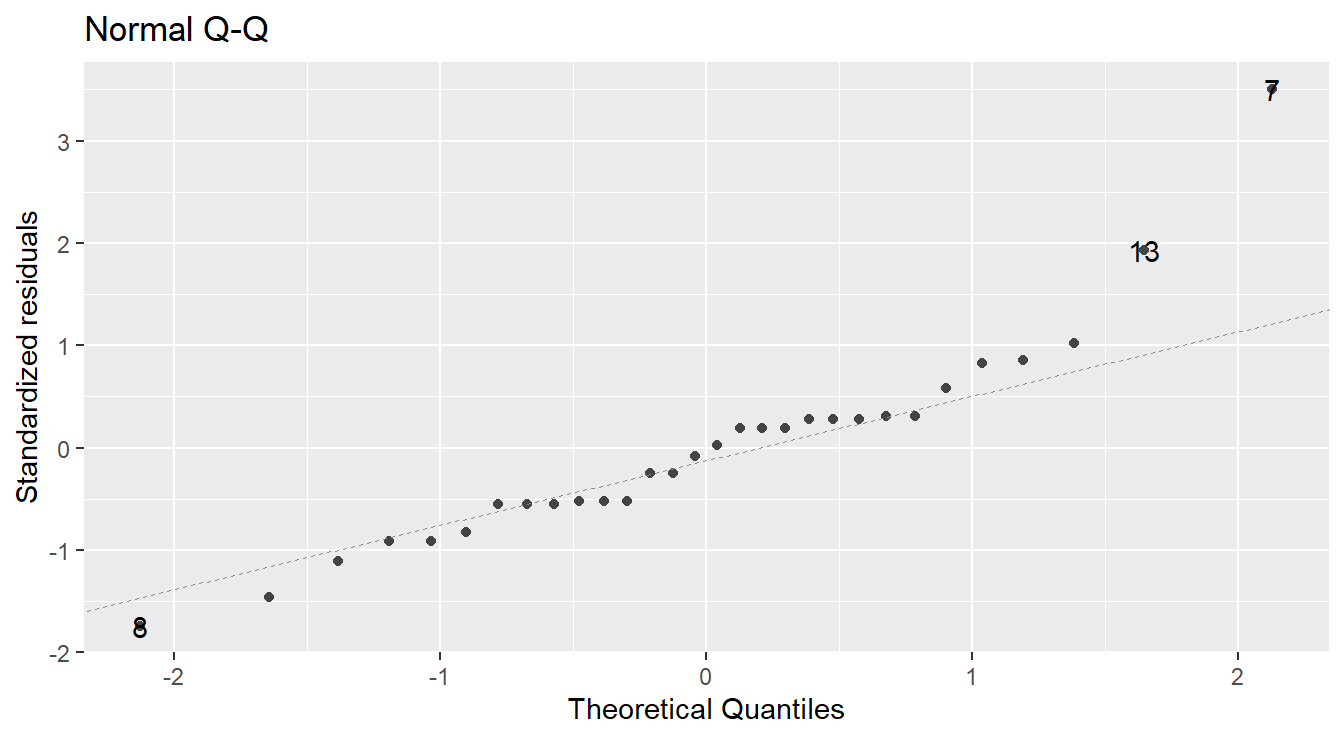
Figure 2.3: Normal Q-Q plot demonstrating the residuals may deviation from the Normality assumption.
In a normal Q-Q plot, the observed sample values (the “sample quantiles”) are plotted against the corresponding quantiles from a reference normal distribution (the “theoretical quantiles”). If the parent population is normally distributed, then the sample values should reasonably match up one-to-one with normal reference quantiles, resulting in a plot of points that line up in a linear “45-degree” fashion.
A normal Q-Q plot of the residuals is presented in Figure 2.3. A common tool to analyze these plots is sometimes known as the “fat pencil” test. Consider the thick pencils children are given when learning to write – lay that pencil on the plotted line, if all the points are covered by the pencil then the data is approximately normally distributed.
In Figure 2.3 there is some deviation from the line (observations 13 and 7). Coupled with the issues with variance outlined above, we may have evidence to suggest the normality assumption is not met.
Note: It is quite common for violations of constant variance and normality to go hand-in-hand. That is, if there are concerns about one, it is likely there will be concerns regarding the other.
2.5.3 Code to check assumptions
The code to generate the above plots can be achieved via the autoplot function when including the ggfortify library (Horikoshi et al., 2019).
The autoplot function on the fitted model provides 4 plots by default as seen in Figure 2.4 (note: we will discuss the Constant Leverage plot in Section 8.6).
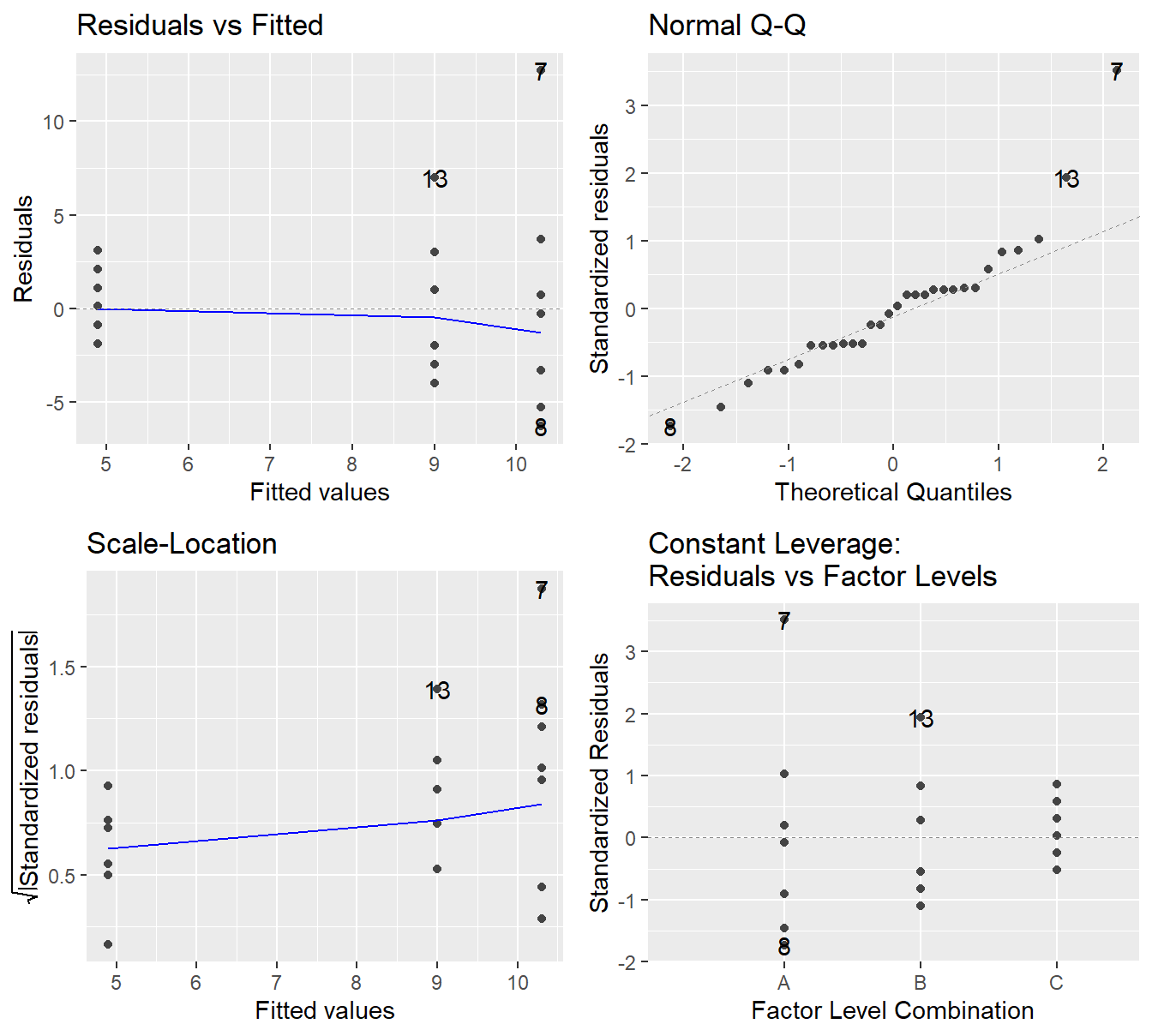
Figure 2.4: Residual plots for the One-Way ANOVA studying the effect of different drugs on immobilization times - Top-left a Residuals vs Fitted Plot; Top-right a Normal Q-Q plot of the residuals; Bottom-left the Scale-Location plot; and Bottom-right a Residuals vs Leverage plot.
2.6 Transforming your response
In cases where we have violations of our underlying assumptions, a common remedy is to transform the response variable.
We have evidence from the above plots that the variance appears to be increasing with the response (determined by assessing the constant variance assumption) and that there may be some violations to normality. One way to typically address this issue is to transform the response variable. We will go into more details later but common transformations include \(Y^*=log_{10}(Y)\), \(Y^*=\sqrt{Y}\) and \(Y^*=1/Y\). Here we will use a logarithmic as it is fairly common. Later in the text we will describe other transformations in more detail. We can achieve this in R with the following which provides the residuals plots in Figure 2.5.
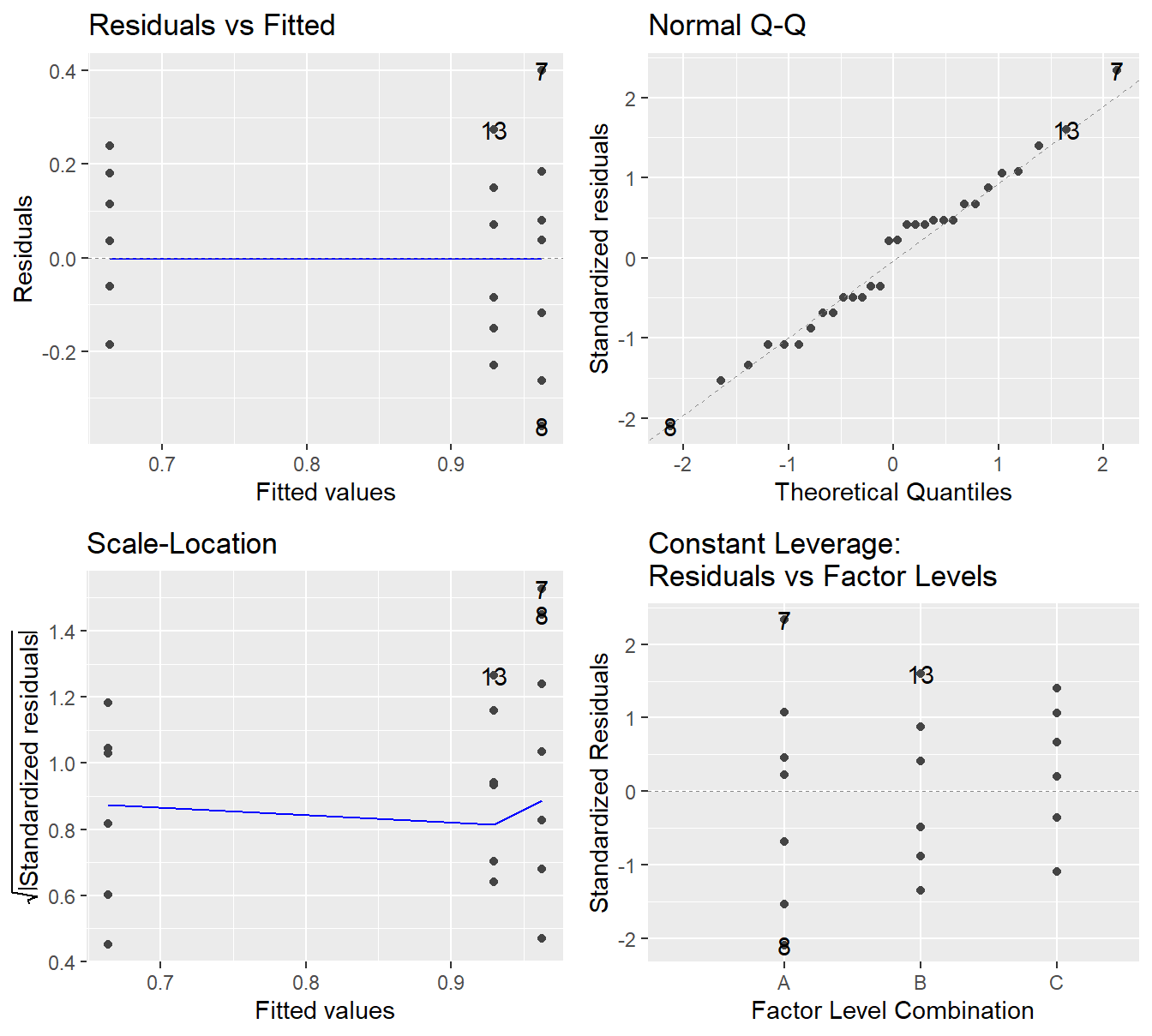
Figure 2.5: Residual diagnostic plots when the response variable is the logarithm of the immobilization time. Here the heteroskedasticity and non-normality in the original model has been improved, albeit not entirely mitigated, with some minor fanning in the residuals vs fitted plot.
Note the improvement in the Normal Q-Q plot – the observed quantiles closely match the theoretical quantiles. The Scale-Location plot shows that the issues with a changing variance have largely been addressed as we see a nearly horizontal line. In the Residuals vs Fitted plot the violation in constant variance has improved although some indication the vertical spread is increasing but is present.
Overall, the logarithmic transformation has improved our model assumptions. We can try other transformations but a perfect transformation is unlikely to exist. When a variance increases with the fitted value, a logarithm on the response tends to useful (and fairly interpretable). Later in the text we will discuss another approach to transforming the response, but for now we proceed with the logarithmic transformation.
Although logarithms tend to be somewhat easy to interpret, we should note that our model has changed. Specifically, if you consider equation (2.3) with regard to our working example, \(Y\) is the knockdown time but we fit the ANOVA model on \(\log_{10}(Y)\). Using the ideas of (2.2), the model can be rewritten as \[\log_{10}(Y_{i}) = \mu_A + \delta_B X_{Bi} + \delta_C X_{Ci} + \varepsilon_{ji}\] where \(\mu_A\) is the mean of group \(A\). \(X_{Bi}\) takes on the value 1 if the \(i^\mathrm{th}\) observation is in group \(B\), otherwise it is zero. Likewise \(X_{Ci}\) for group \(C\). The value \(\delta_B\) measures how the mean of group \(B\) differs from \(A\), and \(\delta_C\) how group \(C\) differs from \(A\).
If we were to “un”-log the above model to the original units by raising each side to the power of 10, we get. \[10^{\log_{10}(Y_i)} = Y_i = 10^{\mu_A + \delta_B X_{Bi} + \delta_C X_{Ci} + \varepsilon_{ji}} = 10^{\mu_A} \times 10^{\delta_B X_{Bi}}\times 10^{\delta_C X_{Ci}} \times 10^{\varepsilon_{ji}}\] by properties of logarithms and power rules. So technically speaking, in terms of the original units, we have fit a multiplicative model.
Interpretation of the multiplicative model is a bit more nuanced than that of a linear model. First, we need to recall a few properties of logarithms and powers from an Algebra course.
- \(\log_{10}(1) = 0\).
- For an \(x\) such that \(0 < x < 1\), \(\log_{10}(x) < 0\).
- For an \(x>1\), \(\log_{10}(x) > 0\).
- \(10^0 = 1\)
- For an \(x < 0\), then \(0 < 10^x < 1\).
- For an \(x > 0\), then \(10^x > 1\)
2.7 Performing ANOVA
In the above, we fit a initial model, assessed its assumptions, transformed the response variable and fit the model again. Specifically, the following code was used.
# Fit the model with the original response
fit.drug <- aov(Knockdown_time ~ Drug, data=deer_knockdown_times)
# Check model assumptions
autoplot(fit.drug)
# We determined a transformation is warranted due to issues
# with the constant variance and normality assumption
fit.log.drug <- aov(log10(Knockdown_time) ~ Drug, data=deer_knockdown_times)
autoplot(fit.log.drug)
# Model assumptions looked acceptable.Now that we have assessed that we reasonably meet the underlying assumption, we can proceed to perform the analysis. The test comparing the three drug population means (on a log scale) is provided via the summary() command:
## Df Sum Sq Mean Sq F value Pr(>F)
## Drug 2 0.5357 0.26786 8.219 0.00163 **
## Residuals 27 0.8800 0.03259
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1The result is significant (\(F\) = 8.219, \(\textrm{df}_1\) = 2, \(\textrm{df}_2\) = 27, \(p\)-value = 0.00163). Thus, we could conclude that at least two of the drugs produce significantly different log-mean immobilization times.
Math Tangent: Note that the logarithm function is a monotone increasing function. So if \(a>b\) then \(\log(a)>\log(b)\). Thus, we can infer that the mean immobilization times for the three drug treatments also differs. Also, recall from an algebra course that the base of the logarithm does not affect the overall behavior of the function (you can easily convert from one base to another with the ‘change of base formula’). For this reason, we recommend you use a base-10 logarithm (coded in R with log10()) as it is commonly used in most scientific fields and is fairly interpretable.
Conclusions of ANOVA
The results of the simple One-Way ANOVA tells us that the logarithm of knockdown time differs among the three drug treatments; essentially it tells us that the null hypothesis \(H_0:\ \mu_A = \mu_B = \mu_C\) is rejected in favor of an alternative. Of course, what remains to be seen is which pairs of drugs produce different log-mean immobilization times. For that, we will need to look at techniques known as multiple comparison procedures.
2.8 Follow-up procedures – Multiple Comparisons
When there is no significant difference detected among treatment means, we can safely conclude that the treatment had no effect on the mean response. This effectively says that any differences in means were no larger than the type of change we might expect just due to random chance. In such a case, the analysis is essentially over, except that we might want to assess the size of the observed treatment differences for descriptive purposes.
However, if significant differences are detected among the treatments (as in the knockdown times example above), the ANOVA \(F\)-test does not inform you where the differences lie. In such a situation, a follow-up procedure known as a multiple comparison procedure is necessary to seek out pairwise mean differences.
There are numerous multiple comparison methods in existence, and they all have certain advantages and disadvantages. Some are designed for specific follow-up comparisons and are customizable (known as contrasts). Here we will discuss procedures for handling the following scenarios:
- All possible pairwise comparisons
- All comparisons versus a control group
So for example, if we had a one-way design with five treatment groups A, B, C, D and E, there would be 10 possible pairwise comparisons:
\[\begin{array}{cccc} A~\textrm{vs.}~B & B~\textrm{vs.}~C & C~\textrm{vs.}~D & D~\textrm{vs.}~E \\ A~\textrm{vs.}~C & B~\textrm{vs.}~D & C~\textrm{vs.}~E & \\ A~\textrm{vs.}~D & B~\textrm{vs.}~E & & \\ A~\textrm{vs.}~E & & & \end{array}\]
whereas if (say) A was a control group and we were only interested in comparisons versus a control group, then there would be only 4 pairwise comparisons of interest:
\[\begin{array}{c} A~\textrm{vs.}~B \\ A~\textrm{vs.}~C \\ A~\textrm{vs.}~D \\ A~\textrm{vs.}~E \\ \end{array}\]
Now, you may say to yourself, “So what is the big deal? I’ll just do 10 two-sample \(t\)-test!”
Well, if you find yourself in a multiple comparison situation, it is because you have declared that at least two groups differ based on the result of a single hypothesis test (the \(F\)-test). For that test, you used a specific criterion to make the decision (probably \(p\)-value < 0.05). You performed the analysis with an accepted false positive rate, known as the Type I error rate, or \(\alpha\).
But to tease out where the differences are, you now need to run multiple tests under the same “umbrella”. That gives rise to something known as the multiple testing problem. The problem is an increase in the false positive rate (the Type I error rate) that occurs when statistical tests are used repeatedly.
Multiple testing problem.
If \(m\) independent comparisons are made, each using a Type I error rate of \(\alpha\), then the family-wise (overall) Type I error rate (FWER) for all comparisons considered simultaneously is given by \[\alpha_{FWER} = 1 - (1 - \alpha)^m.\]
Illustration: Coin flips.
Suppose that we declare that a coin is biased if in 10 flips it landed heads at least 9 times. Indeed, if one assumes as a null hypothesis that the coin is fair, then it can be shown that the probability that a fair coin would come up heads at least 9 out of 10 times is 0.0107. This is relatively unlikely, and under usual statistical criteria such as “reject \(H_0\) if \(p\)-value < 0.05”, one would declare that the null hypothesis should be rejected — i.e., declare the coin is unfair.
A multiple testing problem arises if someone wanted to use this criteria (which is appropriate for testing the fairness of a single coin) to test the fairness of many coins. So, imagine you were to test 100 coins using this method. For any particular coin (i.e., some pre-selected coin), we would expect the likelihood of it flipping heads 9 or 10 times to be relatively small (in fact, it would be with probability 0.0107). However, seeing some coin (as in any of the 100 coins) behave that way, without concern for which one, would be more likely. In fact, the probability that all 100 fair coins are identified as fair by this criterion is \((1 - 0.0107)^{100} \approx 0.34\). This suggest, even if all 100 coins were in fact fair, the probably some coin is incorrectly flagged as unfair is approximately 66%. Therefore the application of a “single coin fairness criterion” to multiple comparisons would more likely than not falsely identify at least one fair coin as unfair!
The above illustration may seem contrived, but multiple testing is a reality and a natural follow-up procedure to ANOVA.
To address the problem, many multiple comparison procedures have been developed that help control the accumulation of the false positive rate.
Some methods perform better than others, but common to all of them is making adjustments to \(p\)-values or CIs based upon how many comparisons are being made.
Multiple comparisons in an ANOVA framework can be performed using the add-on R package emmeans (Lenth, 2019).
The package emmeans is quite powerful. We only demonstrate a few pieces of its functionality here, for those interested in a more in depth look, we suggest checking out the emmeans website.
We will focus on two useful methods: Tukey’s HSD and Dunnett’s.
2.8.1 Tukey’s HSD method
One of the more widely used methods is attributed to statistician John Tukey (creator of the Box-Whiskers plot and credited with creating the word “bit”).
It is referred to as Tukey’s Honestly Significant Difference method (honestly!).
It has the advantage of controlling the Type I error rate when performing all possible pairwise comparisons.
First we load the library and then run our fitted model fit.log.drug through the emmeans() function and provide it a formula to inform how we wish to do the comparisons. In this case the formula is simple since we only have a single factor input ~ Drug. Lastly, to do a “pairwise” comparison via the contrast() function.
## Warning: package 'emmeans' was built under R version 4.4.3## Welcome to emmeans.
## Caution: You lose important information if you filter this package's results.
## See '? untidy'## contrast estimate SE df t.ratio p.value
## A - B 0.0328 0.0807 27 0.406 0.9136
## A - C 0.2984 0.0807 27 3.696 0.0027
## B - C 0.2657 0.0807 27 3.291 0.0076
##
## Results are given on the log10 (not the response) scale.
## P value adjustment: tukey method for comparing a family of 3 estimatesThe significant differences are what we expected based on our earlier EDA: drug C is significantly different in terms of log-mean immobilization time as compared to the other two drugs. This is dertermined from the Tukey-adjusted \(p\)-values of 0.0027 (when comparing drugs A and C) and 0.0076 (when comparing drugs B and C). B and A are not significantly different (\(p\)-value of 0.9136). The \(p\)-values in this output have been adjusted for performing 3 comparisons, so we can just compare them to our usual 0.05.
We can also obtain simultaneous 95% confidence intervals for the true mean differences due to each drug pairing using the confint() function on our contrast of an emmeans object:
## contrast estimate SE df lower.CL upper.CL
## A - B 0.0328 0.0807 27 -0.1674 0.233
## A - C 0.2984 0.0807 27 0.0983 0.499
## B - C 0.2657 0.0807 27 0.0655 0.466
##
## Results are given on the log10 (not the response) scale.
## Confidence level used: 0.95
## Conf-level adjustment: tukey method for comparing a family of 3 estimatesThese may be interpreted as follows:
95% CI for \(\mu_A - \mu_B\) = (-0.167, 0.233). This interval contains 0, so we cannot conclude that the true log-mean immobilization time significantly differ between drugs A and B.
95% CI for \(\mu_A - \mu_C\) = (0.0983, 0.499). We can be 95% confident that drug A produces a true log-mean immobilization time that is between 0.0983 to 0.499 higher than drug C.
95% CI for \(\mu_B - \mu_C\) = (0.066, 0.466). We can be 95% confident that drug B produces a true log-mean immobilization time that is between 0.066 and 0.466 higher than drug C.
You should note in the above output that emmeans provides a warning that the results are provided on a log10 scale. To put back in the original units we could “un”-log the intervals by taking base-10 exponents (that is, raise 10 to the power of the value). When using a logarithmic transformation, emmeans provides the functionality to put the response in the original units.
## contrast ratio SE df lower.CL upper.CL
## A / B 1.08 0.200 27 0.68 1.71
## A / C 1.99 0.370 27 1.25 3.15
## B / C 1.84 0.343 27 1.16 2.92
##
## Confidence level used: 0.95
## Conf-level adjustment: tukey method for comparing a family of 3 estimates
## Intervals are back-transformed from the log10 scaleWe can also visualize these results via a plot:
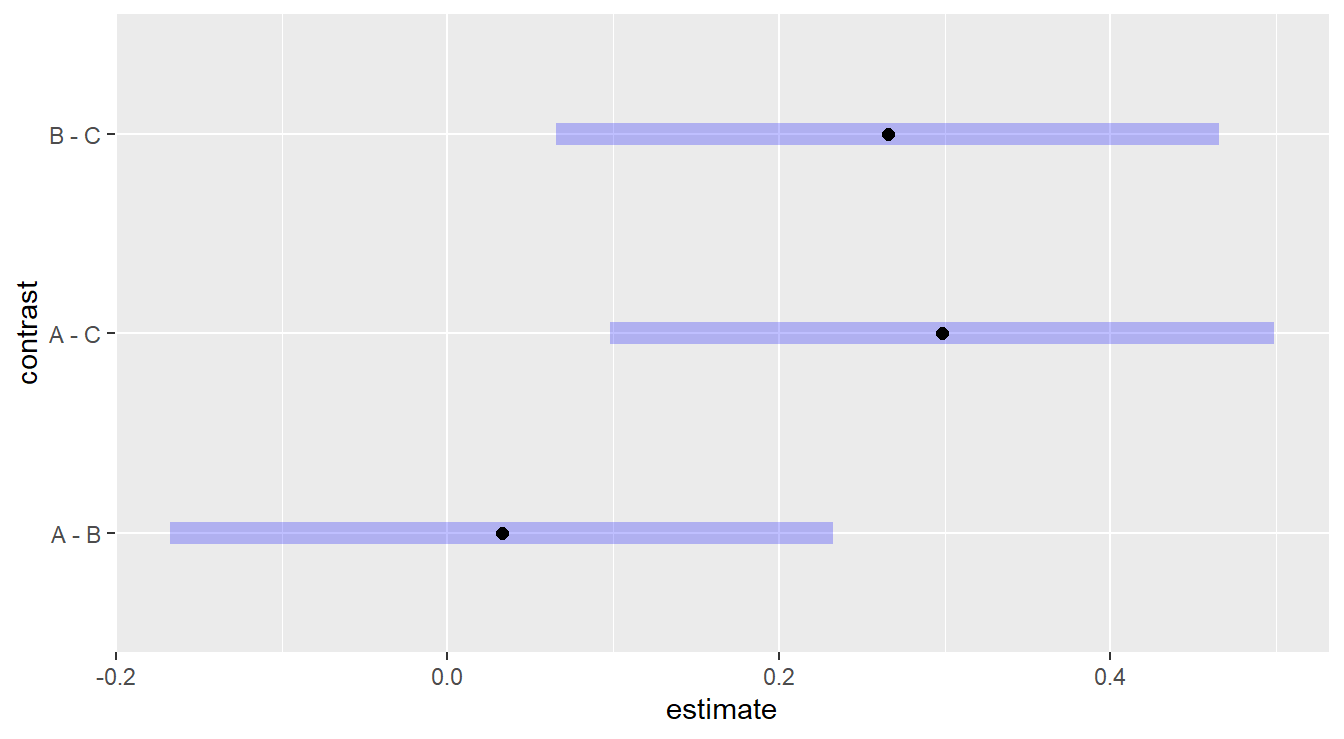
Figure 2.6: Tukey adjusted confidence interval plots comparing treatments treatments A, B and C. Here, we see that treatments B and C are significantly different as well as treatments A and C. We also note there is no significant difference between treatments A and B.
Here, we are interesting in comparing each plotted confident band to the value of 0.
2.8.2 Dunnett multiple comparisons
The Dunnett method is applicable when one of your experimental groups serves as a natural control to which you want to compare the remaining groups.
Note that there is no control group in our working example, but we include the code below for demonstration purposes where treatment level “A” is the reference (ref) control group.
In situations were a control group is present, it is advisable to use a method that only controls the family-wise error rate for the comparisons of interest, rather than over-controlling for all possible comparison such as when using a method like Tukey HSD.
We still use the emmeans package but specify the comparisons are against a control group. The code is below:
## contrast estimate SE df t.ratio p.value
## B - A -0.0328 0.0807 27 -0.406 0.8750
## C - A -0.2984 0.0807 27 -3.696 0.0019
##
## Results are given on the log10 (not the response) scale.
## P value adjustment: dunnettx method for 2 testsThe two comparisons provided are A vs. B and A vs. C. The only significant difference is between A and C (\(p\)-value = 0.0019). As before, here are simultaneous CIs and plots:
## contrast estimate SE df lower.CL upper.CL
## B - A -0.0328 0.0807 27 -0.222 0.157
## C - A -0.2984 0.0807 27 -0.488 -0.109
##
## Results are given on the log10 (not the response) scale.
## Confidence level used: 0.95
## Conf-level adjustment: dunnettx method for 2 estimates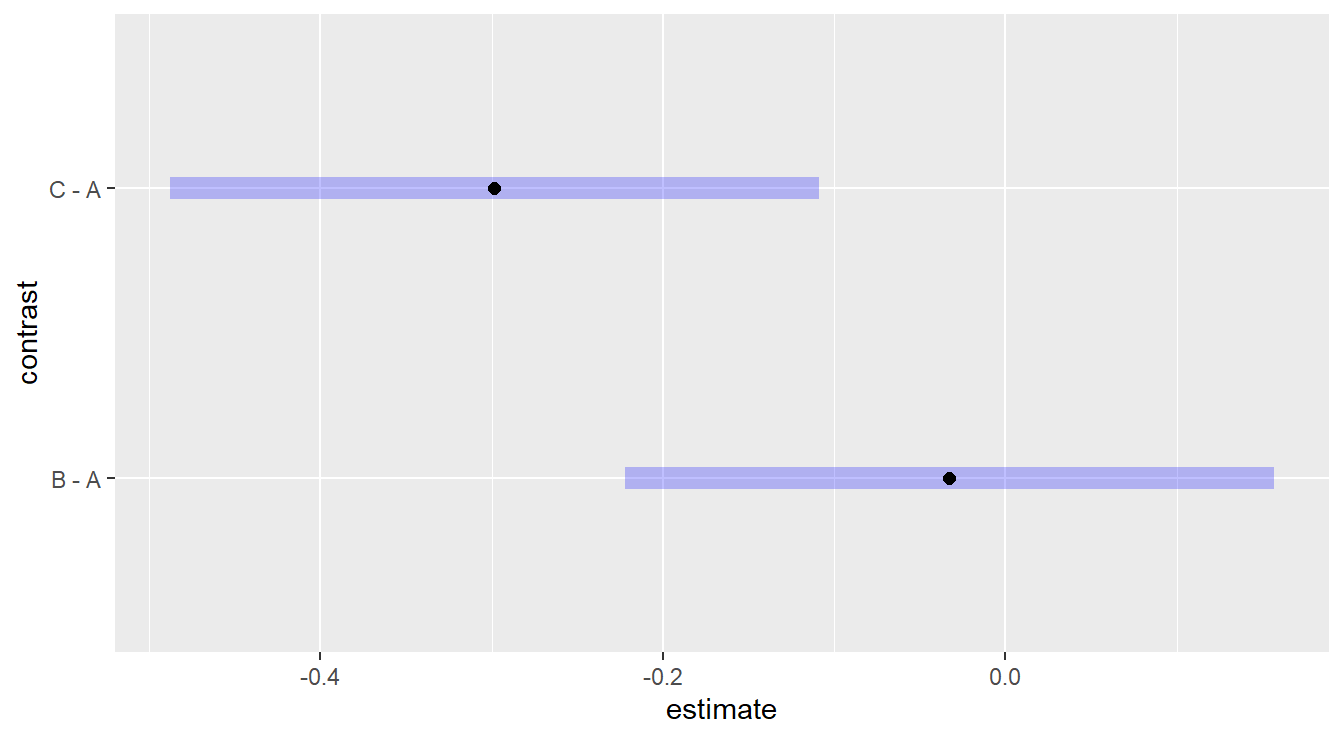
Figure 2.7: Dunnett adjusted confidence interval plots demonstrating that treatment c is significantly different than control group a, while treatment b is not significantly different than control group a.
It is interesting to compare the results you get via the two methods (Tukey vs Dunnett) for the same comparisons (e.g., A vs. C). Can you explain what you see?
Note. If you do not specify a ref= in the contrast function, group *A* will automatically be chosen as the reference group for Dunnett (the default behavior is to choose the first factor level [in this case the first alphabetically]). We recommend you always explicitly tell the code the reference groups. In the below example we set the second (2) factor, *B*, to be the control withref=“B”`.
## contrast estimate SE df t.ratio p.value
## A - B 0.0328 0.0807 27 0.406 0.8750
## C - B -0.2657 0.0807 27 -3.291 0.0054
##
## Results are given on the log10 (not the response) scale.
## P value adjustment: dunnettx method for 2 tests